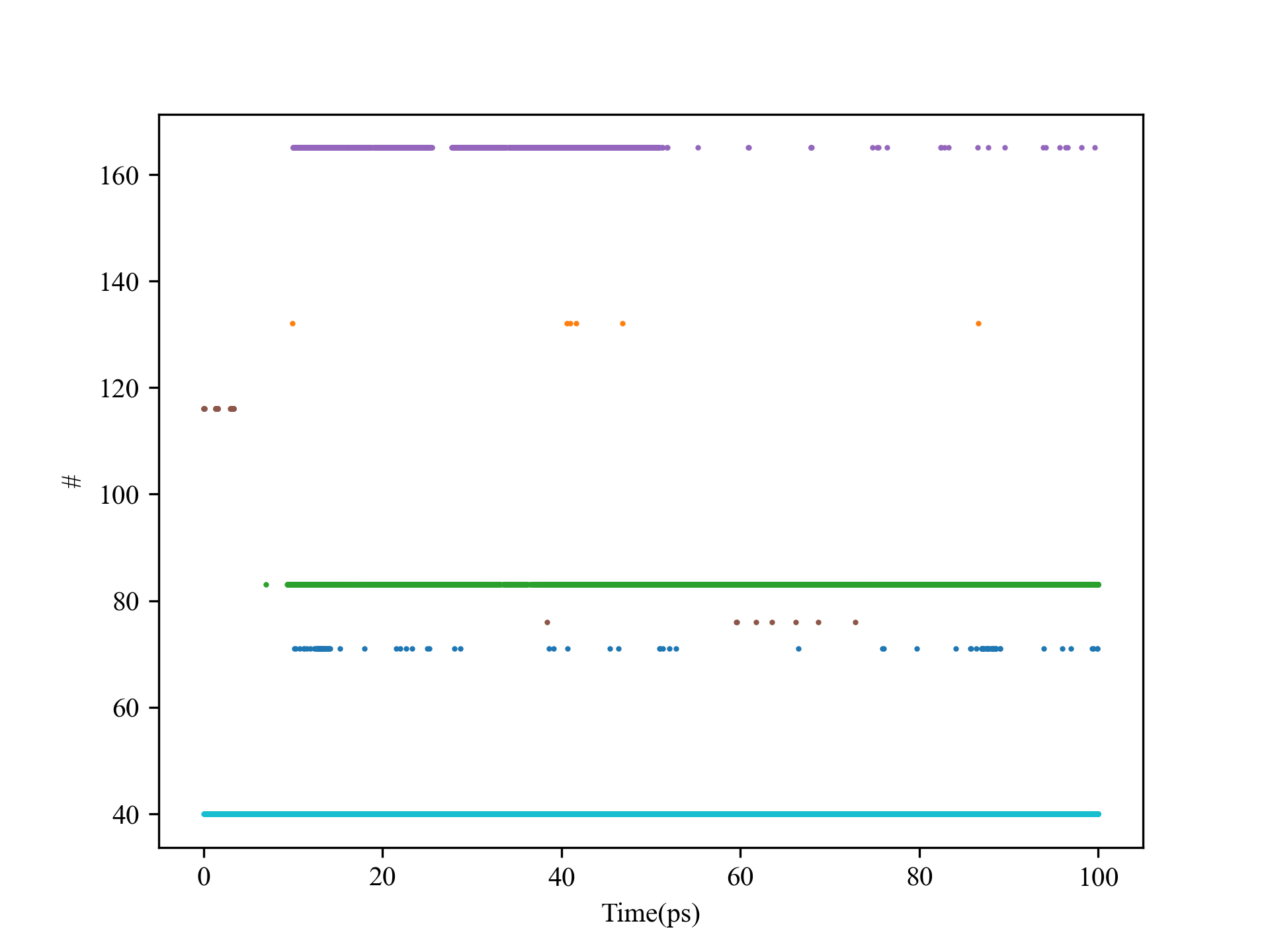
importMDAnalysisasmdafromtqdmimporttqdmimportnumpyasnpimportmatplotlib.pyplotaspltimportargparseplt.rcParams['font.family']='Times New Roman'parser=argparse.ArgumentParser(description="A Python script for calculating the residence time of a certain molecule in the " \
"ion solvation shell within a specific distance from the electrode")parser.add_argument("-gro","--gro",type=str,help="gro file",default="./md.gro")parser.add_argument("-xtc","--xtc",type=str,help="xtc file",default="./md.xtc")parser.add_argument("-ion","--ion",type=str,help="string of the selection of ions")parser.add_argument("-elec","--elec",type=str,help="string of the selection of electrode")parser.add_argument("-sol","--sol",type=str,help="string of the selection of solvate")# 必须是单个原子parser.add_argument("-id","--id",type=float,help="maximum distance of ions from the electrode, unit: Ångström")parser.add_argument("-sd","--sd",type=float,help="maximum distance of solvate from the ion, unit: Ångström")parser.add_argument("-frame","--frame",type=int,help="frame number")parser.add_argument("-dt","--dt",type=float,help="step size, unit: ps")args=parser.parse_args()u=mda.Universe(args.gro,args.xtc)sel_ion=u.select_atoms(f"({args.ion}) and (around {args.id} ({args.elec}))",updating=True)j=0sel_sol=u.select_atoms(f"{args.sol}")result=np.zeros((int(sel_sol.n_atoms),args.frame))foriintqdm(u.trajectory[:args.frame]):sel_ion_index=sel_ion.indicesforid_ioninsel_ion_index:sel_mol=u.select_atoms(f"{args.sol} and (around {args.sd} (index {id_ion}))",updating=True)sel_mol_index=sel_mol.indicesforminsel_mol_index:m+=1# 调整至gro中的原子序号ifm<9095:# 在前100中k=(m-3654)//5else:# 在后100中k=100+(m-9095)//5result[k,j]=1j+=1result[result==0]=np.nannum=result.shape[1]foriinrange(int(sel_sol.n_atoms)):plt.scatter(np.arange(num)*args.dt,result[i]+i,s=1)plt.xlabel("Time(ps)")plt.ylabel("#")plt.savefig("./md.png",dpi=300)